Wiki¶
CR de réunions¶
Réunion du 8/06/2022:¶
Présents: Claire, Yannick, Sarah.
Liens utiles:
https://elearning.formation-permanente.inrae.fr/course/view.php?id=196§ion=1
https://github.com/nf-core/rnaseq/blob/master/docs/output.md
Exemples
MultiQC v1.9- Exemple: http://genoweb.toulouse.inra.fr/~sigenae/sarah/multiqc_report.html
https://genotoul-bioinfo.pages.mia.inra.fr/use-nextflow-nfcore-course/
https://forgemia.inra.fr/genotoul-bioinfo/use-nextflow-nfcore-course
Autres réunions sur GitLab CATI BIOS4BIOL¶
https://forgemia.inra.fr/bios4biol/Help4MultiQC/-/tree/main
Exemples de graphiques commentés¶
General Stats¶
Il y a effectivement une petite différence de comptage entre samtools flagstat et samtools (raw_total_sequences) mais la différence est effectivement liée au multimapping :
or 136.5 + 6.5 = 143
M Total seqs (16ieme ligne) = nb de reads brutes pour l'union R1 + R2 = 136.5
M Non-Primary (11ieme ligne) = endroits où les reads mappent à plusieurs endroits (au minimum 2 mappings) = 6.5
M Reads (1ere ligne) = compte les lignes dans le BAM donc ajoute les multimappées aussi.
Chez toi, ces différences sont grandes car tu as beaucoup de multimapping. Voir pourquoi avec la PF de séquençage ?
Pour la différence proper paired, samtools et rseq, j'ai aussi des résultats très différents , du simple ou double.... d'autant que j'ai très peu de multimaps (5)...
Après, reseq est un vieil outil, utilisé juste pour checker la qualité...
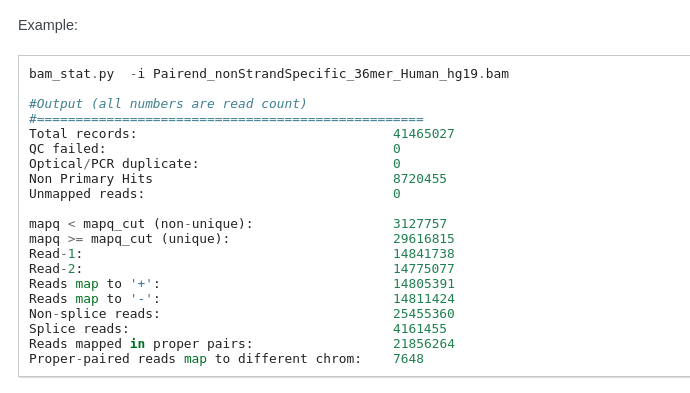
La ligne de commande rseq est:
RNASEQ:RSEQC:RSEQC_BAMSTAT (E_L1_90_LM_M_M+_R1) COMPLETED 0 4m 59s 100.5% 1 MB
bam_stat.py \
-i E_L1_90_LM_M_M+_R1.markdup.sorted.bam \
\
> E_L1_90_LM_M_M+_R1.bam_stat.txt
bam_stat.py --version | sed -e "s/bam_stat.py //g" > rseqc.version.txt
RSeQC v2.3.7
bam_stat.py: Now counts ‘Proper-paired reads map to different chrom’
http://rseqc.sourceforge.net/#bam-stat-py --> Il compte les paires donc dans l'exemple ci-dessous: 41 millions de records mais 21 M de paired donc 50% environ... Donc peut-être que multiQC calcule un % mais sur les mauvais chiffres.... Faudrait comprendre par qui (multiQC ?) est calculé le pourcentage ?
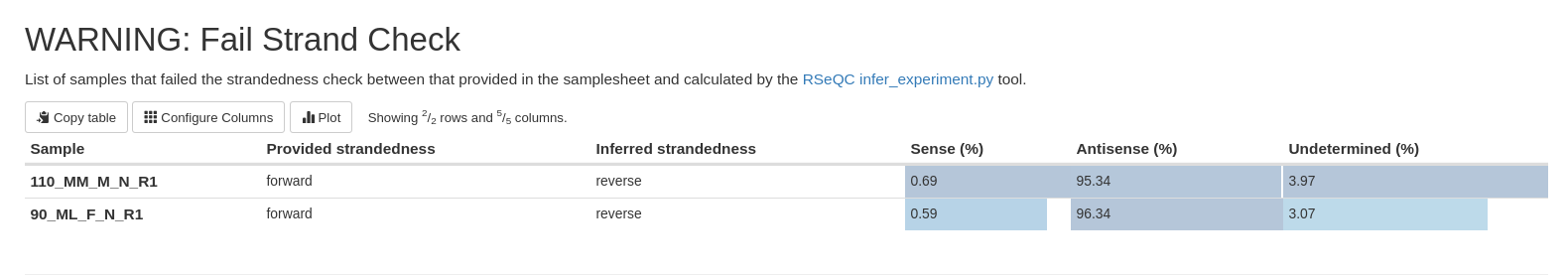
Utiliser Rseq pour vérifier foward/reverse:
Lancer un pipeline sur 2-3 échantillons avec l'indication "unstranded" puis analyser le multiQC report généré par Nextflow nf-core rnaseq version 3.
En effet, RSeQC infer_experiment nous indique le pourcentage de lectures qui mappent en antisense et en sense. Si les librairies sont séquencées à partir de la polyA, l'antisense est le sens 3'-5' donc reverse et le sense est 5'-3' donc forward.
Autre indication toujours à partir du report multiQC, avec l'outil qualimap.
RSeQC¶
2012 - Assez peu utilisé - Contrôle qualité.
FastQC (raw)¶
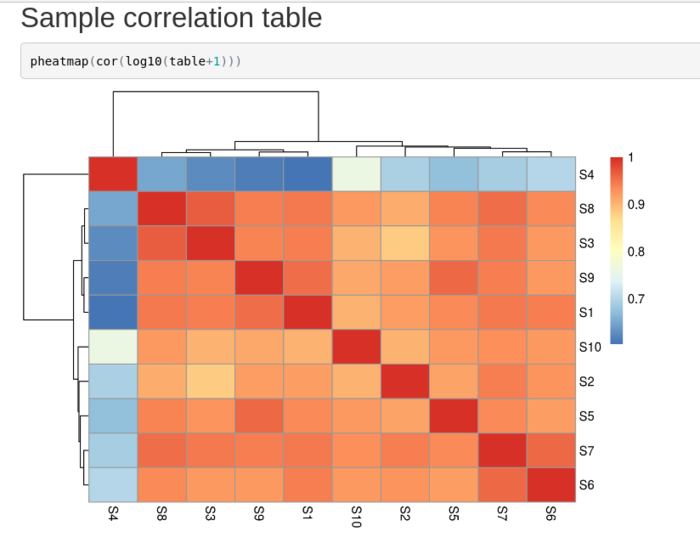
-> Cf e-learning FPN INRAESample correlation¶
sample_correlation : corresponds à une heatmap sous R. Pour mettre en évidence les échantillons outlayers ainsi que la ressemblance des échantillons dans leur expression.
Exemple: s4 est un outlayer
stringtieFPKM¶
Quantification des gènes du modèle Ensembl.
preseq¶
Recherche des lectures brutes redondantes en marquant les lectures identiques. Cette comparaison des lectures aboutit à un tableau avec, en première colonne, le nombre de lectures testées et les 3 autres colonnes sont les
lectures distinctes. Une lecture est comparée par rapport aux autres pour voir si elle est nouvelle ou pas. Cette analyse de la redondance de l'information permet de savoir s'il est nécessaire de continuer à séquencer ou pas.
markDuplicates¶
Marquage des lectures identiques dans le BAM.
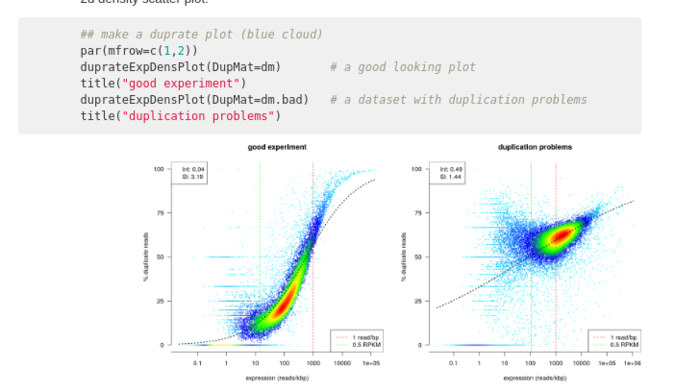
dupradar¶
Recherche les duplicats PCR dans les données afin de déterminer si le lot et l'expression sont biaisés par la duplication des lectures hors du séquençage.
Mini-projets des masters II BioInfo à compiler¶
Voir les pièces jointes ci-dessous. Sujets proposés en 2022 et 2023 à l'ensemble des étudiants du master II BioInfo de l'UPS.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}