Organisation des données utilisateurs¶
- Organisation des données utilisateurs
Les données sont organisées en types structurés.
Les types sont implémentés dans un module contenu dans le package DATA.
Pour utiliser un des types cités ci-dessous, il suffit d'importer le modules m_qtlmap_types du package DATA.
Toutes les subroutines de QTLMap prennent en entrée un objet de type QTLMAP_DATASET qui contient l'ensemble des informations disponibles de l'utilisateur (jeux de données, paramètre analyse et options en ligne de commande)
QTLMAP_DATASET¶
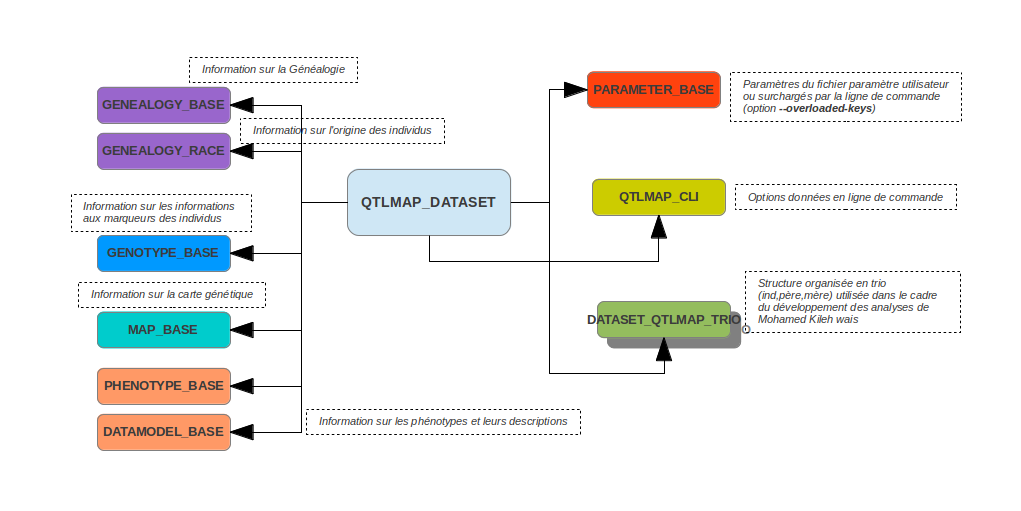
type ,public :: QTLMAP_DATASET type(MAP_BASE) , pointer :: map => null() type(GENEALOGY_BASE) , pointer :: genea => null() type(GENEALOGY_RACE) , pointer :: geneaRace => null() type(GENOTYPE_BASE) , pointer :: genoAnimal => null() type(PHENOTYPE_BASE) , pointer :: phenoAnimal => null() type(DATAMODEL_BASE) , pointer :: phenoModel => null() type(PARAMETER_BASE) ,pointer :: params => null() type(QTLMAP_CLI),pointer :: cli => null() !//New structure used for multitraits analysis (These Mohamed Kileh wais) type (DATASET_QTLMAP_TRIO), public , pointer :: datasetUser contains procedure ,public :: set => set_qtlmap_dataset procedure ,public :: copy => copy_qtlmap_dataset procedure ,public :: release => release_qtlmap_dataset end type QTLMAP_DATASET
Le type QTLMAP_DATASET contient un ensemble d'attribut définit également par un type structuré.
GENEALOGY_BASE¶
Interface¶
le fichier source
type , public :: GENEALOGY_BASE integer :: ngp=0 integer :: ngm=0 integer , dimension (:), pointer :: ngmgp => null() integer , dimension (:), pointer :: nrgm => null() character(len=LEN_DEF) , dimension (:),pointer :: gmere => null() character(len=LEN_DEF) , dimension (:),pointer :: gpere => null() integer :: nr=0 character(len=LEN_DEF) , dimension (:),pointer :: repro => null() character(len=LEN_DEF) , dimension (:),pointer :: reprop => null() character(len=LEN_DEF) , dimension (:),pointer :: reprom => null() integer , dimension (:), pointer :: rep_reprop => null() integer , dimension (:), pointer :: rep_reprom => null() integer , dimension (:), pointer :: ndm => null() integer , dimension (:), pointer :: nmp => null() character(len=LEN_DEF) , dimension (:),pointer :: mere => null() character(len=LEN_DEF) , dimension (:),pointer :: pere => null() character(len=LEN_DEF) , dimension (:),pointer :: animal => null() integer :: nd=0 integer :: nm=0 integer :: np=0 integer :: nfem = 0 integer , dimension (:),pointer :: reppere => null() integer , dimension (:),pointer :: repmere => null() character(len=LEN_DEF) , dimension (:),pointer :: femelle => null() integer , dimension (:),pointer :: repfem => null() character(len=LEN_DEF),dimension(:,:),pointer :: OldGenealogy => null() integer :: OldGenealogySize = 0 contains procedure, public :: copy => copy_genealogy_base procedure, public :: release => release_genealogy_base procedure ,public :: print => print_genealogy_base end type GENEALOGY_BASE
Parcourir la généalogie des F1¶
type(GENEALOGY_BASE) , pointer :: genea ! // Pointeur sur l'objet genea de type GENEALOGY_BASE genea => dataset%genea !// genea%ngp => nombre de grand-père (F0) !// genea%ngm => nombre de grand-mère (F0) !// genea%nr => nombre de reproducteur (F1) do igp=1,genea%ngp do jgm=genea%ngmgp(igp)+1,genea%ngmgp(igp+1) do kr=genea%nrgm(jgm)+1,genea%nrgm(jgm+1) !// kr est le reproducteur (F1) du père indicé par igp et de la mère indicé par jgm !// genea%repro(kr) => nom du reproducteur !// genea%gpere(igp) => nom du père du reproducteur kr !// genea%gmere(jgm) => nom de la mère du reproducteur jgm end do end do end do
- ngm comptabilise la somme des mères intra-famille de grand père. c.a.d un animal "grand-mere", accouplé à deux animaux "grand-père", est comptabilisé 2 fois !
Parcourir la généalogie des F2¶
type(GENEALOGY_BASE) , pointer :: genea ! // Pointeur sur l'objet genea de type GENEALOGY_BASE genea => dataset%genea !// genea%np => nombre de père (F1) !// genea%nm => nombre de mère (F1) !// genea%nd => nombre de descendant (F2) do ip=1,genea%np do jm=genea%nmp(ip)+1,genea%nmp(ip+1) do kd=genea%ndm(jm)+1,genea%ndm(jm+1) !// kd est le descendant (F2) du père indicé par ip et de la mère indicé par jm !// genea%animal(kd) => nom du reproducteur !// genea%pere(ip) => nom du père du descendant kd !// genea%mere(jm) => nom de la mère du descendant kd !// genea%reppere(ip) => indice kr correspondant au reproducteur père !// genea%repmere(jm) => indice kr correspondant au reproducteur mère end do end do end do
- nm comptabilise la somme des mères intra-famille de grand père. c.a.d un animal "mere", accouplé à deux animaux "père", est comptabilisé 2 fois !
- les tableaux
reppere,repmerepermettent de recuperer les indices du tableaureprogenea%repro(genea%reppere(ip))est equivalent àgenea%pere(ip)genea%repro(genea%repmere(jm))est equivalent àgenea%mere(jm)
Parcours la liste des femelles¶
On considère ici qu'une mère peut être accouplée à deux ou plusieurs mâles.
Voici un exemple de code pour parcourir la liste des femelles du jeux de données
!//genea%nfem => nombre de femelle (<= genea%nm) do ifem=1,genea%nfem !// genea%femelle(ifem) => nom de la femelle end do
On peut parcourir la liste des mères (liste des femelles intra-famille de père) pour traiter les femelles du jeu de données
logical , dimension(dataset%genea%nfem) :: actionFemelle actionFemelle = .false. do jm=1,dgenea%nm !// genea%repfem(jm) => indice correspondant au tableau genea%femelle if ( .not. actionFemelle(genea%repfem(jm))) then ... actionFemelle(genea%repfem(jm)) = .true. end if end do
Exemple de parcours¶
- Construction de la matrice d'incidence pour ajouter les effets polygéniques : parcours F2
- Construction de la matrice d'incidence pour ajouter les effets QTL femelles : parcours liste des femelles
Recuperer les informations "brutes" du fichier de généalogie¶
Le tableau OldGenealogy contient les informations textuelles du fichier de généalogie :
OldGenealogySize: le nombre d'enregistrementOldGenealogy(:,1): 1ère colonneOldGenealogy(:,2): 2ème colonneOldGenealogy(:,3): 3ème colonne
GENEALOGY_RACE¶
le fichier source
type , public :: GENEALOGY_RACE character(len=LEN_DEF) , dimension (:),pointer , public :: racep => null() character(len=LEN_DEF) , dimension (:),pointer , public :: racem => null() character(len=LEN_DEF) , dimension (:),pointer , public :: nom_race => null() integer , public :: NB_RACES=1 contains procedure ,public :: copy => copy_genealogy_race procedure ,public :: release => release_genealogy_race end type GENEALOGY_RACE
- Les tableaux
racep,racemsont dimensionnés avec le nombre de reproducteur (genea%nr) et donne la race du reproducteur - Le tableau
nom_raceest dimensionné avec NB_RACES et liste les races de la population étudié - Cette structure de donnée est accessible si l'utilisateur renseigne la clef in_race du fichier p_analyse
do ip=1,genea%np do jm=genea%nmp(ip)+1,genea%nmp(ip+1) do kd=genea%ndm(jm)+1,genea%ndm(jm+1) racep(genea%reppere(ip)) ! // race du pere ip racem(genea%repmere(jm)) ! // race de ma mere jm end do end do end do
PHENOTYPE_BASE¶
type ,public :: PHENOTYPE_BASE character(len=LEN_DEF) , dimension (:),pointer :: bete => null() real(kind=dp) ,dimension(:,:),pointer :: y => null() real(kind=dp) ,dimension(:,:),pointer :: cd => null() integer , dimension (:,:),pointer :: ndelta => null() logical ,dimension (:,:),pointer :: presentc => null() integer ,dimension (:,:),pointer :: nivx => null() real (kind=dp) ,dimension (:,:),pointer :: covar => null() integer ,dimension(:,:) ,pointer :: ydiscretord => null() character(len=LEN_DEF) ,dimension(:,:) ,pointer :: ycategorial => null() integer , dimension (:),pointer :: corperf => null() ! // Contains the corresponding index of y from an index animal ! // Estimabilité / nb de descendant phenotype : (1er dim indicie caractere/ 2 eme dim : indice mere) integer :: ndmin = 0 logical , dimension (:,:),pointer :: estime => null() integer , dimension (:),pointer :: nmumest => null() ! // Number of dam with estimability integer , dimension (:),pointer :: namest => null() ! // Number of female with estimability integer , dimension (:,:) ,pointer :: iam => null() !! // Indexe of the female contains procedure ,public :: copy => copy_phenotype_base procedure ,public :: release => release_phenotype_base end type PHENOTYPE_BASE
Descrition des données¶
- le tableau
betedonne le nom des animaux enregistrés dans le fichier de phénotype - le tableau
yindicé par un individus et un l'indice du caractère donne la valeur du phénotype continue "valeur réelle" - le tableau
ycategorialindicé par un individus et un l'indice du caractère donne la valeur du phénotype discret "valeur caractère" - le tableau
ydiscretordindicé par un individus et un l'indice du caractère donne la modalité du phénotype discret - le tableau
cd???(les données censurées) - le tableau
ndeltaindique si la valeur est manquante (=0) ou présente (=1) - le tableau
presentcindique si la valeur est manquante (=.false.) ou présente (=.true.) - le tableau
nivxindique les niveaux des effets fixés - le tableau
covarindique les valeurs des covariables - la valeur
ndminindique le nombre de progéniture minimum pour étudier la transmission d'un QTL en famille de plein-frère- Cette information devrait répertorié dans la partie "paramètres de l'analyse"
- le tableau
estimede taillegenea%nmindique si une famille de plein frère doit être considérée par l'analyse (dépend dendmin) - le tableau
nmumestdonne le nombre de mère estimable pour un caractère donné(dépend dendmin) - le tableau
namestdonne le nombre de femelle estimable pour un caractère donné(dépend dendmin)
Parcours des performances des F2¶
type(GENEALOGY_BASE) , pointer :: genea type(PHENOTYPE_BASE) , pointer :: dpa genea => dataset%genea dpa => dataset%phenoAnimal !//Pour un caractere indéxé par icar do ip=1,genea%np do jm=genea%nmp(ip)+1,genea%nmp(ip+1) do kd=genea%ndm(jm)+1,genea%ndm(jm+1) !// si la valeur de la performance du caractere ic est présente pour l'individu kd if (dpa%presentc(icar,kd)) then !//performance !//dpa%y(icar,kd) !//dpa%cd(icar,kd) ... end if end do end do end do
DATAMODEL_BASE¶
type ,public :: DATAMODEL_BASE integer , dimension (:,:),pointer :: modele => null() integer :: ncar = 0 integer :: ncarcat = 0 character(len=LEN_DEF) , dimension (:),pointer :: carac => null() character(len=1) , dimension (:),pointer :: natureY => null() integer , dimension(:),pointer ,public :: filter_car => null() integer ,dimension(:,:) ,pointer :: indicemod => null() !// Correspond value from a original discrete value and the array ydiscretord integer ,dimension(:) ,pointer :: nmod => null() !// Number of classes corresponding with discrete value real(kind=dp) ,dimension(:,:) ,pointer :: seuil => null() !// Thresholds on the underlying scale real(kind=dp) ,dimension(:,:) ,pointer :: prop => null() !// Proportion of the discrete classes integer :: ncov = 0 character(len=LEN_DEF) , dimension (:),pointer :: namecov => null() integer :: nfix = 0 character(len=LEN_DEF) , dimension (:),pointer :: namefix => null() integer , dimension (:), pointer :: nlev => null() character(len=LEN_DEF) , dimension (:,:) ,pointer :: listelev => null() real (kind=dp), dimension (:) , pointer :: h2 => null() real (kind=dp), dimension (:,:) , pointer :: RhoP => null() real (kind=dp), dimension (:,:) , pointer :: RhoG => null() real (kind=dp), dimension (:) , pointer :: xmut => null() real (kind=dp), dimension (:) , pointer :: sigt => null() type(model_trait) , dimension(:),pointer :: listModelTrait => null() contains procedure ,public :: copy => copy_datamodel_base procedure ,public :: release => release_datamodel_base procedure ,public :: write_file => write_datamodel_base end type DATAMODEL_BASE
Description des données¶
Ces informations proviennent du fichier modèle
- le tableau
modele:modele(ic,1)=> nombre d'effets fixés dans le modèle pour le caractereicmodele(ic,2)=> nombre de covariables dans le modèle pour le caractereicmodele(ic,3)=> nombre d'effets fixés en interaction avec le QTL dans le modèle pour le caractereicmodele(ic,3+ifix)=> pourifix <= modele(ic,1), donne l'indice dans la liste des effets fixés pour le caractereicmodele(ic,3+nfix+icov)=> pouricov <= modele(ic,2), donne l'indice dans la liste des covariables pour le caractereicmodele(ic,3+nfix+ncov+ifix)=> pourifix <= modele(ic,3), donne l'indice de l'effet en interaction avec le QTL dans la liste des effets fixés pour le caractereic
Attention, cette structure de données ne modelise pas l'interaction avec un n-ieme autre qtl dans le modèle!
- ncar : nombre de caractère
- ncarcat : nombre de caractère de type catégoriel
- le tableau
caracdonne le nom de chaque caractère
- le tableau natureY donne la nature de chaque carectere
- "i" : discret ordonée (--calcul=2 et --calcul=21)
- "c" : catégoriel (Il n'existe pas d'analyse QTL pour ce type d'analyse)
- "r" : donnée réelle (majorité des analyses développés)
*filter_carest un tableau qui a permis de sélectionner les caractères à analyser lors de la lecture du fichier modèle parmi les phénotypes enregistrés. L'utilisateur donne cet liste juste après la descriptions des modèles pour chaque variable caractère dans le fichier modèle.
- nfix : nombre d'effets fixés
- le tableau
namefixdonne le nom de chaque effets fixés - le tableau
nlev(ifix)donne le nombre de niveau pour l'effet fixéifix - le tableau
listelev(ifix,il)donne la chaîne de caractère (définit dans le fichier performance) du niveau il pour l'effet fixé ifix
- ncov : nombre de covariables
- le tableau
namecovdonne le nom de chaque covariable
- le tableau
h2(ic)donne l'héritabilité du caractèreic - le tableau
RhoP(ic,jc)donne la corrélation phénotypique entre les caractèresicetjc - le tableau
RhoG(ic,jc)donne la corrélation génotypique entre les caractèresicetjc - le tableau
xmut(ic)donne la moyenne des valeurs enregistrées pour le caractèreic - le tableau
sigt(ic)donne l'écart type des valeurs enregistrées pour le caractèreic
Les données de type discret ordonné¶
- le tableau
indicemod(ic,id)donne l'indice de chaque modalité du caractèreic- le tableau est trié avec comme premiere élément : la modalité la moins représenté (plus petit effectif) et dernier élément, la modalité la plus représenté (le plus grand effectif)
- le tableau
nmod(ic)donne le nombre de modalité/classe pour le caractèreic - le tableau
prop(ic,imod)donne l'effectif en pourcentage de la modalitéimodet du caractereicsum(prop(ic,:)) = 1
- le tableau
seuil(ic,imod)donne le quantile associé (fonction de distribution cumulative pourx<=imod)
Le type model trait¶
Le type structuré DATAMODEL_BASE contient un tableau de typetype(model_trait). Cette structure est une structure contenant de l'information redondante vis à vis le tableau modèle.Le tableau modèle, comme le type
type(model_trait), informe pour chaque caractère :
- le nombre d'effets fixés, le nombre de covariables et le nombre d effets fixés en interaction avec un QTL intervenant dans le modèle.
Le type structuré type(model_trait) permet de définir également les effets fixés en interaction avec les autres QTL pour les calculs utilisant l'option --qtl>=2.
type , public :: model_trait !// number of fixed effect integer :: nbfe !// number of covariate integer :: nbco !// number of qtl with interaction integer :: nbqtlinter !// number of interaction with the qtl for each qtl with interaction defined, size : nbqtlinter integer ,dimension(:) ,pointer :: nbint => null() !// size : nbfe : reference integer ,dimension(:) ,pointer :: indexFixedEffect => null() !// size : nbco : reference integer ,dimension(:) ,pointer :: indexCovariate => null() !//size : nbqtlinter,nbint integer ,dimension(:,:) ,pointer :: indexFixedEffectWithInteraction => null() contains procedure, public :: release => release_model_trait procedure, public :: copy => copy_model_trait end type model_trait
listModelTrait(ic)%nbfeest nombre d'effets fixés intervenant dans le modèle du caractèreiclistModelTrait(ic)%indexFixedEffect(ifix)est l'indice duifixeffet fixé intervenant dans le modèle du caractèreic
listModelTrait(ic)%nbcoest nombre de covariables intervenant dans le modèle du caractèreiclistModelTrait(ic)%indexCovariate(icov)est l'indice duicovcovariable intervenant dans le modèle du caractèreic
listModelTrait(ic)%nbqtlinterest nombre d'effets fixés en interaction avec un QTL intervenant dans le modèle du caractèreiclistModelTrait(ic)%indexFixedEffectWithInteraction(iqtl,ifix)est l'indice duifixéffet fixé en interaction avec leiqtlQTL intervenant dans le modèle du caractèreic
MAP_BASE¶
type MAP_BASE real , private :: BASE_STEP = 1.d2 ! //Step analysis integer , public :: PAS = 0 ! //Number of chromosome to analyse integer , public :: nchr = 0 ! //List of chromosome name character(len=LEN_S) ,dimension(:) , pointer, public :: chromo => null() ! //Number of the marker by chromosome integer , dimension (:), pointer, public :: nmk => null() ! //Name of the markers by chromosome character(len=LEN_DEF), dimension (:,:), pointer,public :: mark => null() ! //Average position in Morgan by chromosome and marker real (kind=dp), dimension (:,:), pointer,public :: posi => null() ! //Mal position in Morgan by chromosome and marker real (kind=dp), dimension (:,:), pointer,public :: posim => null() ! //Female position in Morgan by chromosome and marker real (kind=dp), dimension (:,:), pointer,public :: posif => null() ! //Xaldane distance computed with posim position real (kind=dp), dimension (:,:,:), pointer,public :: rm => null() ! //Xaldane distance computed with posif position real (kind=dp), dimension (:,:,:), pointer,public :: rf => null() ! //get abscis in Morgan corresponding to a a position N on the chromosome CHR real (kind=dp) ,dimension(:,:),pointer ,public :: absi => null() contains procedure, public :: copy => copy_map_base procedure, public :: link => link_map_base procedure, public :: release => release_map_base procedure, public :: get_maxnpo procedure, public :: set_base_and_step procedure, public :: get_long_step_morgan procedure, public :: set_absi procedure, public :: get_ilong procedure, public :: get_dx procedure, public :: get_pos procedure, public :: get_npo procedure, public :: get_flanking_marker procedure, public :: print => print_map_base end type MAP_BASE
Description des données¶
nchrnombre de chromosome pris en compte pour l'analyse des données- le tableau
chromo(ich)donne le nom du chromosomeich - le tableau
nmk(ich)donne le nombre de marqueur porté par le chromosome ich - le tableau
mark(ich,ll)donne le nom du marqueurllporté par le chromosomeich - le tableau
posi(ich,ll)donne la position en Morgan (carte consensus) du marqueurllporté par le chromosomeich - le tableau
posim(ich,ll)donne la position en Morgan (carte mâle) du marqueurllporté par le chromosomeich - le tableau
posif(ich,ll)donne la position en Morgan (carte femelle) du marqueurllporté par le chromosomeich - le tableau
rm(ich,ll1,ll2)donne la distance en haldane (carte mâle) entre les marqueurll1etll2portés par le chromosomeich - le tableau
rf(ich,ll1,ll2)donne la distance en haldane (carte femelle) entre les marqueurll1etll2portés par le chromosomeich - le tableau
absi(ich,ix)donne la position en Morgan d'un indiceix(échantillonnage pour l'interval mapping)
Échantillonnage du groupe de liaison¶
L'utilisateur fournit un "pas" d'étude (exemple opt_step=0.05 pour une analyse tout les 5 cMorgans)
BASE_STEPest la valeur (de type entier) tel queBASE_STEP * opt_step > 0PAS = BASE_STEP * opt_step
BASE_STEP = 10**3 = 1000PAS = 5
Le nombre de positions échantillonnées à testé sur le groupe de liaison (GL) porté par le chromosome chr:
ilong=(nint(map%BASE_STEP*(map%posi(chr,map%nmk(chr))-map%posi(chr,1))) / map%pas) + 1
Les fonctions membres¶
get_maxnpofournit le nombre maximum de position à tester sur un chromosome - Cette fonction est utilisée pour allouer un tableau indexé sur l'échantillonnage du GL (une courbe de LRT par exemple)get_ilong(ich)donne le nombre maximum de position qu'on peut tester sur le chromosomeichget_dx(ich,ix)donne la valeur en Morgan de l'index positionixsur le chromosomeichget_pos(ich,pos)donne l'indiceixd'une position en Morgan porté par le chromosomeichget_npo(ich)donne le nombre de position à tester sur un GL (utiliser pour le parcours du GL)get_flanking_marker(chr,ix,flleft,flright)retourne dansflleftetflrightl'index des marqueurs flanquants de l'index position ix
Exemple de parcours des groupes de liaison¶
type(QTLMAP_DATASET) ,intent(in) :: dataset type(MAP_BASE) , pointer :: map integer :: chr,npo map => dataset%map allocate (lrt(map%nchr,map%get_maxnpo())) do chr=1,map%nchr do ipo=1,map%get_npo(chr) !//calcul du LRT lrt(chr,ipo) = .... end do end do
GENOTYPE_BASE¶
type ,public :: GENOTYPE_BASE integer , dimension (:,:),allocatable ,public :: nall character(len=LEN_DEF) , dimension (:,:,:),allocatable ,public :: alleles real (kind=dp), dimension (:,:,:), allocatable ,public :: pc_all integer :: nmes = 0 character(len=LEN_DEF) , dimension (:),pointer :: numero => null() integer(kind=KIND_PHENO),dimension (:,:,:,:),pointer :: pheno => null() character(len=LEN_DEF) , dimension (:,:) ,pointer :: value_pheno => null() integer , dimension (:) ,pointer :: nb_value_pheno => null() integer , dimension (:),pointer :: corregp => null() integer , dimension (:),pointer :: corregm => null() integer , dimension (:),pointer :: correr => null() integer , dimension (:),pointer :: correm => null() integer , dimension (:),pointer :: correp => null() integer , dimension (:),pointer :: corred => null() logical , dimension (:,:), pointer :: presentg => null() logical , dimension (:),pointer :: estfem => null() !// Estimabilité / nb de descendant genotypé ( 1ere dim : indice female) !// Genotype code for missing value integer(kind=KIND_PHENO) , public :: NMANQUE contains procedure ,public :: copy => copy_genotype_base procedure ,public :: release => release_genotype_base procedure ,public :: print => print_genotype_base procedure ,public :: init_pheno procedure ,public :: end_pheno procedure ,public :: set_pheno procedure ,public :: get_pheno procedure ,public :: get_ind_pheno !procedure ,public :: write_file => write_typ end type GENOTYPE_BASE
Description des données¶
nmesest le nombre d'animaux référencé dans le fichier contenant les génotypages- le tableau
numero(id)donne le nom de l'animalidréférencé dans le fichier de génotypage
Codification et Gestion des genotypages dans QTLMap¶
Afin d'optimiser la place mémoire et accélérer les traitements, nous stoquons les données de genotypage dans des entiers codés sur 1 ou 2 octets.- le codage sur 1 octet permet de stoquer 256 valeurs d'allèle
- le codage sur 2 octets permets de stoquer 65535 valeurs d'allèle
le codage sur 1 octet posait des problèmes pour les jeux de données de type microsat (256 valeurs n'etait pas suffisant)
définition des constantes dans le fichier source:trunk/src/data/m_qtlmap_constant.f95
integer ,parameter ,public :: KIND_PHENO = 2 integer(kind=KIND_PHENO) ,parameter ,public :: VAL_MIN_INDEX_PHENO=1-(2**bit_size(VAL_MIN_INDEX_PHENO))/2 integer(kind=KIND_PHENO) ,parameter ,public :: VAL_MAX_INDEX_PHENO=(2**bit_size(VAL_MIN_INDEX_PHENO))/2 - 1
La fonction membre set_pheno permet d'associer un allèle à un entier si celui-ci n'est pas référencé.
Cette fonction retourne le codage (entier sur KIND_PHENO octets) de l'allèle.
value_pheno et nb_value_pheno sont utilisé pour ce codage :
- le tableau
nb_value_pheno(ich)donne le nombre de codage enregistré pour le chromosome ich - le tableau
value_pheno(ich,i)stoque la valeur (chaîne de caractère) du ième codage. ce ième est la valeur de l'entier codé surKIND_PHENOoctets
- La fonction membre
get_phenoretourne la valeur 'chaîne de caractère' d'un entier codé surKIND_PHENOoctets
Utilisation de l'information aux marqueurs chez la population étudiée¶
Le tableau pheno stoque l'ensemble de l'information génétique du jeu de données
- le tableau
pheno(c,ll,id,1)correspond au codage du 1er allèle de l'individuidsur le marqueurllporté par le chromosomec - le tableau
pheno(c,ll,id,2)correspond au codage du 2eme allèle de l'individuidsur le marqueurllporté par le chromosomec - la valeur
nmanquecorrespond au codage d'un allèle manquant
type(GENOTYPE_BASE) , pointer :: dga dga => dataset%genoAnimal do c=1,dataset%map%nchr do ll=1,dataset%map%nmk(c) do ip=1,dataset%genea%np !// Test si les informations aux marqueurs sont disponibles pour le pere ip if ( dga%correp(ip) /= INT_NOT_DEFINED ) then !// test si le premiere ou le deuxieme allele du pere ip au marqueur ll porté par le chromosome c !// est une valeur manquante if (dga%pheno(c,ll,dga%correp(ip),1) == dga%nmanque .or. dga%pheno(c,ll,dga%correp(ip),2) == dga%nmanque) then !//...traitement ... end if end if end do end do end do
Pour afficher la valeur réelle du phenotype marqueur d'un individu il faut utiliser la fonction membre
get_pheno (un print du tableau pheno donnes les entiers codés sur KIND_PHENO octets)
!// Affichage des deux alleles du pere ip au marqueur ll porté par le chromosome c print *,trim(get_pheno(dga,c,dga%pheno(c,ll,dga%correp(ip),1))),' ',trim(get_pheno(dga,c,dga%pheno(c,ll,dga%correp(ip),2)))
Correspondance avec la généalogie¶
- le tableau
corregp(igp)donne l'indice correspondant aux tableauxnumero,phenoetpresentgpour le grand-pèreigp - même principe pour
corregm,correr,correp,corremetcorred INT_NOT_DEFINEDest la valeur manquante => l'information génétique n'existe pas pour l'individu- le tableau
presentg(ich,kd)retourne faux si toute l'information génétique pour l'individu kd sont des valeurs manquantes(dga%presentg(ich,kd)=.not. (all(dga%pheno(ich,:,corred(kd),:)==dataset%genoAnimal%nmanque)))
Pour tester la presence des génotypes, il faut utiliser :
- un test avec@== INT_NOT_DEFINED@ pour les F0 et F1 (corregp,corregm,correr,correp,correm)
- le tableau
presentgpour les F2
type(GENEALOGY_BASE) , pointer :: genea type(GENOTYPE_BASE) , pointer :: dga type(MAP_BASE) , pointer :: map genea => dataset%genea dga => dataset%genoAnimal map => dataset%map do c=1,map%nchr do ll=1,map%nmk(c) do ip=1,genea%np !// si l'information aux marqueurs existe pour le pere ip if ( dga%correp(ip) /= INT_NOT_DEFINED ) then !//traitement ... end if do jm=nmp(ip)+1,nmp(ip+1) do kd=ndm(jm)+1,ndm(jm+1) !// si l'information aux marqueurs existe pour l'individu kd if ( presentg(c,kd) ) then ! // Affiche le premier allele de l'individu kd du marqueur ll porté par le chromosome c print *, dga%get_pheno(dga%pheno(c,ll,dga%corred(kd),1)) end if end do!//kd end do!//jm end do !//ip end do!//ll end do!//c
Information sur la population à étudier¶
- le tableau
nall(ich,ik)donne le nombre d'allèle du marqueurikporté par le chromosomeich - le tableau
alleles(ich,ik,il)donne la chaîne de caractère (valeur définit dans le fichier de typage) de l'allèleildu marqueurikporté par le chromosomeich - le tableau
pc_all(ich,ik,il)donne la fréquence de l'allèle il du marqueurikporté par le chromosomeich
FILES_DATASET¶
On stoque dans ce type tous les noms de fichiers (inputs/outputs).
Ce type est définit dans le module source:trunk/src/data/m_qtlmap_type_dataset.f95
type ,public :: FILE_DATASET logical ,public :: genotypeFileDefined = .false. !// flag to test if a user genotype file existe !logical ,public :: simulMap = .false. !// flag to activate a simulation map logical ,public :: mapFileDefined = .false. !// flag to test if a user map file existe !logical ,public :: simulTraits = .false. !// flag to activate a simulation trait logical ,public :: traitsFileDefined = .false. !// flag to test if a user phenotypic file existe !logical ,public :: simulGenea = .false. !// flag to activate a simulation genealogy logical ,public :: raceFileDefined = .false. !// flag to test if a user breed file existe logical ,public :: geneaFileDefined = .false. !// flag to test if a user genealogy file existe character(len=LENGTH_MAX_FILE) , public :: in_genea='' !// Genealogy file name. character(len=LENGTH_MAX_FILE) , public :: in_race='' !// breed file name. character(len=LENGTH_MAX_FILE), dimension(MAX_FILES_INPUT) , public :: in_perfs='' !// Phenotypic records user files / traits data character(len=LENGTH_MAX_FILE) , public :: in_param_ef='' !// Model files describing fixed effect, covariate, !// interaction fixed effect and qtl for each traits character(len=LENGTH_MAX_FILE) , public :: in_typage='' !// Genotype records user files character(len=LENGTH_MAX_FILE) , public :: in_carte ='' !// File map name character(len=LENGTH_MAX_FILE) , public :: in_parsim = '' !// simulation user file character(len=LENGTH_MAX_FILE), public :: resul='' !// main result file character(len=LENGTH_MAX_FILE), public :: resp='' !// sire result file character(len=LENGTH_MAX_FILE), public :: resm='' !// dam result file character(len=LENGTH_MAX_FILE), public :: pateff='' !// files with Sire QTL effect estimations character(len=LENGTH_MAX_FILE), public :: mateff='' !// files with Dam QTL effect estimations character(len=LENGTH_MAX_FILE), public :: pdedf='' !// Generate file with grand parental segment transmission marginal probabilities character(len=LENGTH_MAX_FILE), public :: pdecouple='' !// Generate file with grand parental segment transmission joint probabilities character(len=LENGTH_MAX_FILE), public :: coeffda='' !// linear combinaison of discriminant analysis output character(len=LENGTH_MAX_FILE), public :: grid2qtl='' !// grid LRT for 2 qtl character(len=LENGTH_MAX_FILE), public :: summary='' !// summary result file character(len=LENGTH_MAX_FILE), public :: sum_2qtl='' !// summary result file 2 qtl character(len=LENGTH_MAX_FILE), public :: simula='' !// fichier de resultat de simulation character(len=LENGTH_MAX_FILE), public :: out_phases='' !// fichier avec les phases completes character(len=LENGTH_MAX_FILE), public :: out_haplotypes='' !// fichier avec les haplotypes character(len=LENGTH_MAX_FILE), public :: out_freqall='' !// allele frequency character(len=LENGTH_MAX_FILE), public :: file_informativity='' !// informativity file about marker and sire on dataset contains procedure ,public :: copy => copy_file_dataset procedure ,public :: release => release_file_dataset end type FILE_DATASET
PARAMETER_BASE¶
Les paramètres de l'analyse qui peuvent être surchargés dans le fichier p_analyse par l'utilisateur.
Ce type est définit dans le module source:trunk/src/data/m_qtlmap_type_parameter.f95
Les autres modules accèdes à ces paramètres via la donnée membre params et les attributs que composent cet objet.
if (dataset%params%ndmin) then
...
end if
ou bien en utilisant des mots clefs définit dans le module source:/trunk/src/data/m_qtlmap_type_parameter.f95#L28
if (dataset%params%key_exist(K_GENEA)) then
file=dataset%params%get_file_val(K_GENEA)
...
end if
Pour plus de détail (créer une nouvelle option du fichier paramètre, utiliser cette option,...) voir la section m_qtlmap_type_parameter.f95.
type ,public :: PARAMETER_BASE
!// number maximum of evaluation (module optimization)
integer , public :: optim_maxeval = 0
!// maxtime for each optimization (module optimization)
real(kind=dp) , public :: optim_maxtime = 0
!// tolerance for the variable x (module optimization)
real(kind=dp) , public :: optim_tolx = 0
!// tolerance for the function evaluation result f (module optimization)
real(kind=dp) , public :: optim_tolf = 0
!// tolerance for the gradient (module optimization)
real(kind=dp) , public :: optim_tolg = 0
!// Precision gradiant df/dx = f(x+h)-f(x-h)/2*h (module optimization)
real(kind=dp) , public :: optim_H_PRECISION = 5.d-5
!// Estimabilité / nb de descendant phenotype : (1er dim indicie caractere/ 2 eme dim : indice mere)
integer , public :: NDMIN = -1
!// option de traitement des familles en demi germains ou melange plein/ demi germains
integer , public :: OPT_SIB = -1
!// Threshold to check the equilibrium of marker transmission within each family
real (kind=dp) , public :: PSEUILHWE
!// threshold of the test to identify an abnormal recombination rate
real (kind=dp) , public :: PROB_SEUIL_RECOMB
!// The analysis is interrupted if for a sire, none of its phases reach this threshold
real (kind=dp) , public :: PHPSEUIL
!// Threshold above which the probable maternal phases will be considered in the analysis
real (kind=dp) , public :: PRSEUIL
!// Cholesky threshold to build X'X matrix
real (kind=dp) , public :: SEUIL_CHO
!// Minimal probability to consider a predicted phase using flanking phased markers
real (kind=dp) , public :: PROB_PHASE_DESC=95.d-2
!// Threshold to test confusion betwwen level inside a contingence matrix
real(kind=dp) , public :: THRES_CONFUSION
!// Threshold for convergence in the linear mode heteroscedastic
real (kind=dp) , public :: EPS_LINEAR_HETEROSCEDASTIC
!// Maximum iteration in the linear mode heteroscedastic to avoid infinity loop
integer , public :: MAX_LINEAR_ITERATION
!// get the id of the gpu device to used
integer , public :: gpu_device_id = 0
!// Minimal gamete probability (LDA Haplotype option)
real(kind=dp) , public :: prob_seuil_gam = 0.25d0
!// Number maximum of haplotype allowed
integer , public :: NB_HAPLO_PRIOR = 0
!// miminum probability of a haplotype in the LD analysis
real (kind=dp) , public :: PROB_HAPLO_MIN = 0.0d0
!
integer , public :: LONG_MIN_IBS = 0
!// Length of the haplotypes to be followed in LD analyses
integer , public :: LONGHAP = 0
!// array of values initialized from generic keys
character(len=LEN_L) , dimension(MAXNB_KEY_DEV_GEN) ,public :: tabDevKey
logical , public :: simul ! the context is a simulation or not
character(len=LENGTH_MAX_FILE), public :: analyse_file ! analyse file
!!// The values of the p_analyses file (referenced by the array keys)
character(len=LENGTH_MAX_FILE) , dimension(:), private ,pointer :: values
!!// The keys defined of the p_analyses file
character(len=LEN_L) , dimension(:), private ,pointer :: keys
contains
procedure ,public :: set => set_parameter_base !// init the object
procedure ,private :: read_analyse_file !// init keys and values arrays
procedure ,private :: check_unknown_keys
procedure ,private :: initialize_dev_generic_keys
procedure ,public :: get_string_val
procedure ,public :: get_int_val
procedure ,public :: get_real_val
procedure ,public :: get_file_val
procedure ,public :: get_summary_panalyse
procedure ,public :: key_exist
procedure ,public :: help_panalyse
procedure ,public :: link => link_parameter_base
procedure ,public :: copy => copy_parameter_base
procedure ,public :: release => release_parameter_base
end type PARAMETER_BASE
Il y a deux structures qui contiennent l'ensembles des informations :
keys: les clefs définis par l'utilisateur dans le fichiers paramètrevalues: la valeurs des clefs définis par l'utilisateur dans le fichiers paramètre
QTLMAP_CLI¶
Ce type est définit dans le module source:trunk/src/data/m_qtlmap_type_cli.f95
Il définit l'ensemble des options que l'utilisateur peut donner en ligne de commande.
type , public :: QTLMAP_CLI !!// default simulation number if is not specified by the user integer ,private :: NSIM_DEFAULT = 1000 !!// default hypothesis number (qtl) to test if is not specified by the user integer ,private :: NQTL_DEFAULT = 1 !!// default value of the family option (full sib family is the default). integer ,private :: SIB_DEFAULT = 2 !!// default optimization method using by qtlmap ( LA optimized analysis ) integer ,private :: OPTIM_DEFAULT = 1 !!// /* OPTION NAME */ !!// calcul option name, see m_qtlmap_analyse module to have the available analysis character(len=LEN_OPT) ,private :: OPT_CALCUL ='--calcul' !!// hypothesis option name, some analysis have limitation with this value. character(len=LEN_OPT) ,private :: OPT_QTL ='--qtl' !!// haplotype option name, see m_qtlmap_haplotype/haplotype to see the availables haplotypes versions character(len=LEN_OPT) ,private :: OPT_HAPLOTYPE ='--haplotype' !!// family option name character(len=LEN_OPT) ,private :: OPT_SIBF ='--family' !!// optimization option name character(len=LEN_OPT) ,private :: OPT_OPTIM ='--optim' !!// random simulation option name character(len=LEN_OPT) ,private :: OPT_SIM ='--simulation' !!// number of simulation option name character(len=LEN_OPT) ,private :: OPT_NSIM ='--nsim' !!// expression phenotypics values option name character(len=LEN_OPT) ,private :: OPT_EQTL ='--data-transcriptomic' !!// permutation simulation option name character(len=LEN_OPT) ,private :: OPT_PERMUT ='--permute' !!// snp option name character(len=LEN_OPT) ,private :: OPT_SNP ='--snp' !!// haplotype dam option name character(len=LEN_OPT) ,private :: OPT_HDAM ='--hdam' !!// biallelic option character(len=LEN_OPT) ,private :: OPT_BIQ ='--biq' !!// interaction between 2 qtls option character(len=LEN_OPT) ,private :: OPT_INTERACTION ='--interaction' !!// censored data option name, Used to take care about a third generation with trait character(len=LEN_OPT) ,private :: OPT_CENSORED_CALCUL ='--calcul-cd' !!// threshold estimation option name character(len=LEN_OPT) ,private :: OPT_ESTIME_THRESHOLDS = '--estimate-thresholds' !!// threshold estimation short option name character(len=LEN_OPT) ,private :: OPT_ESTIME_THRESHOLDS_SHORT = '-s' !!// quiet output console option name character(len=LEN_OPT) ,private :: OPT_QUIET ='--quiet' !!// verbose output console option name character(len=LEN_OPT) ,private :: OPT_VERBOSE ='--verbose' !!// debug output console option name character(len=LEN_OPT) ,private :: OPT_DEBUG ='--debug' !!// quiet output console short option name character(len=LEN_OPT) ,private :: OPT_QUIET_SHORT ='-q' !!// verbose output console short option name character(len=LEN_OPT) ,private :: OPT_VERBOSE_SHORT ='-v' !!// debug output console short option name character(len=LEN_OPT) ,private :: OPT_DEBUG_SHORT ='-d' !!// used to print a classical output analysis in a simulation case or expressions phenotypics values analysis. character(len=LEN_OPT) ,private :: OPT_PRINT_CLASSIC ='--print-all' !!// help parameter file option name character(len=LEN_OPT) ,private :: OPT_HELP_PANALYSE = "--help-panalyse" !!// help simulation parameter file option name character(len=LEN_OPT) ,private :: OPT_HELP_PARAMSIM = "--help-paramsim" !!// help option name character(len=LEN_OPT) ,private :: OPT_HELP ='--help' !!// suppprime l'estimation des effets polygeniques character(len=LEN_OPT) ,private :: OPT_NOPOLY ='--nopoly' !!// overload keys defined in the parameter file character(len=LEN_OPT) ,private :: OPT_OVERLOADED_KEYS ='--overloaded-keys' !!// do not estime Qtl sire effect (useful for analyse of female hetero-chromosme) character(len=LEN_OPT) ,public :: DISABLE_SIRE_QTL ='--disable-sire-qtl' !!// confidence intervalle option name option name character(len=LEN_OPT) ,public :: OPT_CI ='--ci' !!// number of simulation for the bootstrap option to calculate Confidence Intervalle character(len=LEN_OPT) ,public :: OPT_CI_NSIM ='--ci-nsim' contains !// Print QtlMAP Version procedure,public :: cli_print_qtlmap_version !// Print help procedure,public :: cli_print_help !// True if the user ask help message procedure,public :: cli_is_help !// Get the analyse parameters file procedure,public :: cli_get_parameters_file !// Get the analyse type procedure,public :: cli_get_analyse !// Get the Haplotype version procedure,public :: cli_get_haplotype !// Get the Family version procedure,public :: cli_get_family !// Get the optimization procedure,public :: cli_get_optimization !// True if the user want estimate rejection threholds of the current analysis procedure,public :: cli_is_estimate_thresholds !// True if the user ask a simulation procedure,public :: cli_is_simulation !// True if the user ask a transcriptomic analysis/simulation procedure,public :: cli_is_transcriptomic_data !// True if the user ask a permutation method for the simulation procedure,public :: cli_is_permute !// True if the user ask to estimate haplotype dam in the model LD and derivated LD analysis procedure,public :: cli_is_hdam !// True if the user ask to estmate a biallelic qtl procedure,public :: cli_is_biq !// True if the user ask to estmate an qtl in interaction procedure,public :: cli_is_interaction !// True if the user ask an iteration and so a simulation procedure,public :: cli_is_nsim !// Get the number of the simulation procedure,public :: cli_get_nsim !// Get the number of the qtl to detect procedure,public :: cli_get_nqtl !// True if the user ask none information procedure,public :: cli_is_quiet !// True if the user ask verbose information procedure,public :: cli_is_verbose !// True if the user ask to print all information procedure,public :: cli_is_print_all !// True if the user ask debug information procedure,public :: cli_is_debug !// True if the user ask help p_analyse information procedure,public :: cli_is_help_panalyse !// True if the user ask help paramsim information procedure,public :: cli_is_help_paramsim !// True if the user want used the F3 phenotypes procedure,public :: cli_is_calcul_cd procedure,public :: cli_get_overloaded_keys procedure,public :: release => release_qtlmap_cli procedure,public :: key_exist procedure,public :: get_key_list_values procedure,public :: get_key_value procedure ,public :: copy => copy_qtlmap_cli end type QTLMAP_CLI
On peut obtenir les information de la ligne de commande à partir de l'objet dataset
exemple, dans la subroutine analyse :
subroutine analyse(dataset,spt,opt_calcul,opt_qtl,lrtsol,listincsol,rhoi,opt_sim) type(QTLMAP_DATASET) ,intent(in) :: dataset type(PDD_BUILD) ,intent(in) :: spt integer , intent(in) :: opt_calcul,opt_qtl integer ,intent(in) :: opt_sim type(TYPE_LRT_SOLUTION) , intent(inout) ,dimension(dataset%phenoModel%ncar,opt_qtl) :: lrtsol type(TYPE_INCIDENCE_SOLUTION) ,intent(inout) ,dimension(dataset%phenoModel%ncar,opt_qtl+1) :: listincsol real (kind=dp) ,dimension(dataset%phenoModel%ncar,dataset%phenoModel%ncar), intent(out) :: rhoi logical :: biq,interaction logical :: hdam integer :: ic logical :: car_real integer :: type_model = MASK_INIT_MOD_INC type(type_effect_contingence) :: cont_eff hdam = dataset%cli%cli_is_hdam() biq = dataset%cli%cli_is_biq() interaction = dataset%cli%cli_is_interaction() ...
DATASET_QTLMAP_TRIO¶
L'objet dataset contient un objet de type DATASET_QTLMAP_TRIO utilisé dans le cadre des développement des analyses de mohamed kileh wais.
Ces analyses sont multi-caractères et utilisait les phénotypes des F3 pour l'étude d'analyse QTL.
voire les analyses multi-caractères développé dans m_qtlmap_incidence_multi.f95
et la génération des cd et corrélation entre cd m_qtlmap_type_dataset_trio.f95
type DATASET_QTLMAP_TRIO integer :: na = 0 !// number of animal (the population) character(LEN=LEN_DEF) ,dimension(:) ,pointer :: listNA => null() !// the key ID => Name for each animal of the population type(QTLMAP_TRIO) ,dimension(:) ,pointer :: trioKd => null() !// List of all trio parent link integer ,dimension(:) ,pointer :: corranim => null() integer ,dimension(:,:),pointer :: NCK => null() !// nombre de descendant ayant une perf par KD pour le Caracteres i integer ,dimension(:,:,:),pointer :: NCiCjK => null() !// nombre de descendant ayant une perf par KD pour le Caracteres i et j real(kind=dp) ,dimension(:,:,:),pointer :: corcd => null() contains !//Ajoute un trio Pere,Mere,Desc dans la structure generale dataset procedure, public :: add_animal_genea !//Ajoute les performance et l info donne manquante pour chaque Kd procedure, public :: init_perf_animal !//Donne la liste des descendants d'un Individu procedure, public :: get_listProgenies !// Construit l'esperances des performances pour chaque KD de generation 2 ainsi que les CD !// initilise Nck et Nc1c2k procedure,public :: calcul_y_cd procedure,public :: calcul_corcd procedure ,public :: copy => copy_qtlmap_dataset_trio procedure ,public :: release => release_qtlmap_dataset_trio end type DATASET_QTLMAP_TRIO
Description des données¶
nanombre d'animaux dans la population- le tableau
listeNA(id)donne la correspodance avec le nom de l'animalid - trioKd enregistre dans un type structuré, l'information de parenté directe d'un animal ainsi que ces performances
- le tableau
corranimdonne la correspondance pour la tableauanimal( les indexes utilisé par la structureGENEALOGY_BASE) NCK(ic,kd): nombre de progéniture (F3) de l'animalkd(F2) ayant une performance indéxé paricNCiCjK(kd,ic,jc): nombre de progéniture (F3) de l'animalkd(F2) ayant exactement deux performances indéxés paricetjc- corcd(ic,jc,kd) : correlation de CD entre les caractere
icetjcpour l'individukd
type QTLMAP_TRIO !// max trait for QTLMAP_TRIO type description integer :: idSire = -1 !// sire integer :: idDam = -1 !// dam integer :: idKd = -1 !// Kd real(kind=dp) ,dimension(MAXCAR) :: perfKd = 0.d0 logical ,dimension(MAXCAR) :: presentc =.false. end type QTLMAP_TRIO